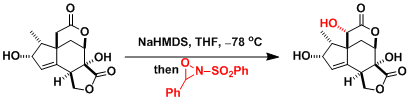
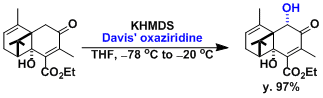
Davis Oxidation
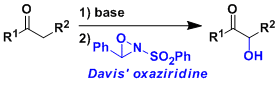
用2-sulfonyloxaziridine (戴維斯試劑)處理烯基氧負離子,羰基和酯基的α位能在溫和條件下被氧化。用樟腦磺酸衍生物之類的具有光學活性的oxaziridine能實現不對稱氧化。
反應機理
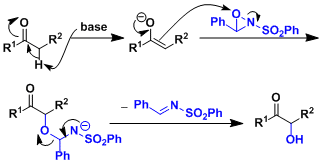
反應實例
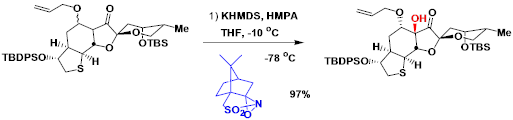
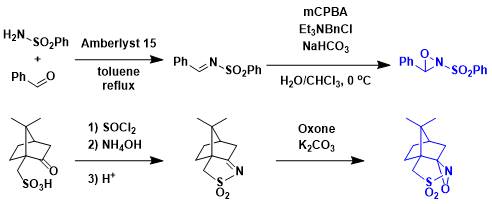
Davisoxaziridine的製備法
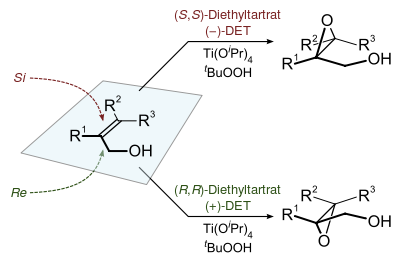
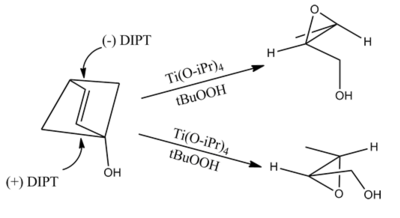
Sharpless epoxidation
The Sharpless epoxidation reaction is an enantioselective chemical reaction to prepare 2,3-epoxyalcohols from primary and secondary allylic alcohols. The oxidizing agent is tert-butyl hydroperoxide. The method relies on a catalyst formed from titanium tetra(isopropoxide) and diethyl tartrate.
5–10 mol% of the catalyst is typical. The presence of 3Å molecular sieves (3Å MS) is necessary.
The epoxidation of allylic alcohols is a well-utilized conversion in fine chemical synthesis. The chirality of the product of a Sharpless epoxidation is sometimes predicted with the following mnemonic. A rectangle is drawn around the double bond in the same plane as the carbons of the double bond (the xy-plane), with the allylic alcohol in the bottom right corner and the other substituents in their appropriate corners. In this orientation, the (−) diester tartrate preferentially interacts with the top half of the molecule, and the (+) diester tartrate preferentially interacts with the bottom half of the molecule. This model seems to be valid despite substitution on the olefin. Selectivity decreases with larger R1, but increases with larger R2 and R3 (see introduction).
The Sharpless epoxidation can also give kinetic resolution of a racemic mixture of secondary 2,3-epoxyalcohols. While the yield of a kinetic resolution process cannot be higher than 50%, the enantiomeric excess approaches 100% in some reactions.

The Sharpless epoxidation is viable with a large range of primary and secondary alkenic alcohols. Furthermore, with the exception noted above, a given dialkyl tartrate will preferentially add to the same face independent of the substitution on the alkene.To demonstrate the synthetic utility of the Sharpless epoxidation, the Sharpless group created synthetic intermediates of various natural products: methymycin, erythromycin, leukotriene C-1, and (+)-disparlure.

The main drawback of this protocol is the necessity of the presence of an allylic alcohol. The Jacobsen epoxidation, an alternative method to enantioselectively oxidise alkenes, overcomes this issue and tolerates a wider array of functional groups.
Jacobsen epoxidation
The Jacobsen epoxidation, sometimes also referred to as Jacobsen-Katsuki epoxidation is a chemical reaction which allows enantioselective epoxidation of unfunctionalized alkyl- and aryl- substituted alkenes. It is complementary to the Sharpless epoxidation (used to form epoxides from the double bond in allylic alcohols). The Jacobsen epoxidation gains its stereoselectivity from a C2 symmetric manganese(III) salen-like ligand, which is used in catalytic amounts. The manganese atom transfers an oxygen atom from chlorine bleach or similar oxidant. The reaction takes its name from its inventor, Eric Jacobsen, with Tsutomu Katsuki sometimes being included. Chiral-directing catalysts are useful to organic chemists trying to control the stereochemistry of biologically active compounds and develop enantiopure drugs.



Katsuki's catalysts
The degree of enantioselectivity depends on numerous factors, namely the structure of the alkene, the nature of the axial donor ligand on the active oxomanganese species and the reaction temperature. Cyclic and acyclic cis-1,2-disubstituted alkenes are epoxidized with almost 100% enantioselectivity whereas trans-1,2-disubstituted alkenes are poor substrates for Jacobsen's catalysts but yet give higher enantioselectivities when Katsuki's catalysts are used. Furthermore, the enantioselective epoxidation of conjugated dienes is much higher than that of the nonconjugated dienes.
The enantioselectivity is explained by either a "top-on" approach (Jacobsen) or by a "side-on" approach (Katsuki) of the alkene.
he mechanism of the Jacobsen–Katsuki epoxidation is not fully understood, but most likely a manganese(V)-species (similar to the ferryl intermediate of Cytochrome P450) is the reactive intermediate which is formed upon the oxidation of the Mn(III)-salen complex. There are three major pathways. The concerted pathway, the metalla oxetane pathway and the radical pathway. The most accepted mechanism is the concerted pathway mechanism. After the formation of the Mn(V) complex, the catalyst is activated and therefore can form epoxides with alkenes. The alkene comes in from the "top-on" approach (above the plane of the catalyst) and the oxygen atom now is bonded to the two carbon atoms (previously C=C bond) and is still bonded to the manganese metal. Then, the Mn–O bond breaks and the epoxide is formed. The Mn(III)-salen complex is regenerated, which can then be oxidized again to form the Mn(V) complex.

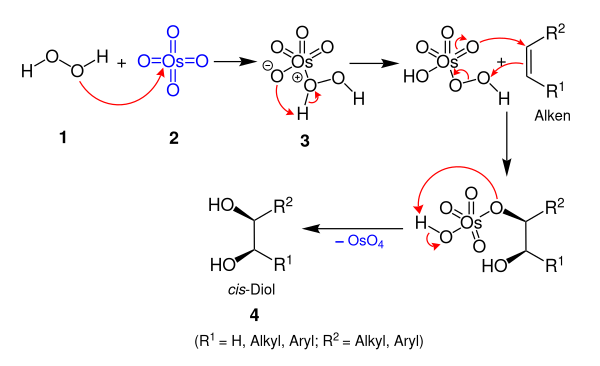
Upjohn Dihydroxylation (OsO4 oxidation)
The Upjohn dihydroxylation is an organic reaction which converts an alkene to a cis vicinal diol. It was developed by V. VanRheenen, R. C. Kelly and D. Y. Cha of the Upjohn Company in 1976. It is a catalytic system using N-methylmorpholine N-oxide (NMO) as stoichiometric re-oxidant for the osmium tetroxide. It is superior to previous catalytic methods.

Prior to this method, use of stoichiometric amounts of the toxic and expensive reagent osmium tetroxide was often necessary. The Upjohn dihydroxylation is still often used for the formation of cis-vicinal diols; however, it can be slow and is prone to ketone byproduct formation. One of the peculiarities of the dihydroxylation of olefins is that the standard "racemic" method (the Upjohn dihydroxylation) is slower and often lower yielding than the asymmetric method (the Sharpless asymmetric dihydroxylation).
In response to these problems, Stuart Warren and co-workers[employed similar reaction conditions to the Sharpless asymmetric dihydroxylation, but replacing the chiral ligands with the achiral quinuclidine to give a racemic reaction product (assuming an achiral starting material is employed). This approach takes advantage of the fact that when using the Sharpless alkaloid ligands, the dihydroxylation of alkenes is faster and higher yielding than in their absence. This phenomenon became known as "ligand accelerated catalysis", a term coined by Barry Sharpless during the development of his asymmetric protocol.
The toxic and volatile OsO4 can also be prepared in situ by the oxidation of K2OsO2(OH)4 with NMO. NMO is also the cooxidant that enables the use of a catalytic amount of OsO4, because this reagent is able to reoxidize an Os(VI) species to an Os(VIII) species:

The mechanism is simplified, for example in alkaline solutions, the catalyst is indeed hydrated.
The key step is the cycloaddition of OsO4 to the olefin. There has been some speculation regarding the actual addition step, for which experimental data suggest the possible involvement of two separate steps. Thus, the question arises during these discussions of whether the key step takes place via an initial (3+2)-addition (1,3-dipolar cycloaddition), or by a (2+2)-addition followed by expansion of the metallacycle.


Quantum chemical calculations have shown an initial (3+2)-addition of OsO4 to be energetically more favorable. However, this energy difference is substantially smaller in the related Re(VII) oxide additions.
Tertiary amines such as DMAP and pyridine accelerate the addition reaction.

Milas hydroxylation
The Milas hydroxylation is an organic reaction converting an alkene to a vicinal diol, and was developed by Nicholas A. Milas in the 1930s.The cis-diol is formed by reaction of alkenes with hydrogen peroxide and either ultraviolet light or a catalytic osmium tetroxide, vanadium pentoxide, or chromium trioxide.

The reaction has been superseded in synthetic chemistry by the Upjohn dihydroxylation and later by the Sharpless asymmetric dihydroxylation.
The proposed mechanism for the Milas hydroxylation involves the initial combination of hydrogen peroxide and the osmium tetroxide catalyst to form an intermediate, which then adds to the alkene, followed by a cleavage that forms the product and regenerates the OsO4.